Introduction
In the 1980s, the pharmaceutical industry had begun expanding beyond domestic markets of individual countries, thanks to globalization. Legislations regarding quality standards of drugs were becoming more complex the world over, and it was becoming difficult for a manufacturer located in one country to match the specific drug regulatory requirements of another country to which they wished to export medicines. Regulators of one country were focused on their regulations and any products that didn’t meet their requirements were rejected. Considering the increasingly international nature of the pharmaceutical market, drug regulators decided to work in collaboration, and this led to the birth of the International Conference on Harmonization (ICH).

ICH and its purpose
Table of Contents
The full form of ICH is the “International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use.” This body was set up to bring together representatives of the pharmaceutical industry and regulatory bodies to discuss technical and scientific aspects of the registration of drugs. As the pharmaceutical industry grew more international, the differences in technical requirements across countries meant that drug makers had to spend a lot of time and money to duplicate test procedures if they wanted to market their products at an international level. It started becoming important to make safe and effective drugs available to patients all over the world without the delays caused by regulations not matching across regulatory bodies of the different countries. Thus, a need was felt to rationalize and harmonize drug regulations, and this resulted in the inception of the International Conference on Harmonization (ICH) in 1990.
The purpose of ICH may be summarized as follows:
- Ensuring quality, safety, and efficacy of drugs.
- Harmonization of drug technical requirements.
- Avoid duplication of human clinical trials.
- Reduce use of animal testing but without a compromise on evaluating efficacy and safety of drugs.
History of ICH
The European Commission pioneered the harmonization concept of pharmaceuticals in the 1980s as it moved towards developing a single market. Observing the success of this, discussions began between the United States, Europe, and Japan to explore the possibility of harmonization.
During the World Health Organization’s Conference of Drug Regulatory Authorities in 1989 in Paris, an action plan was drawn up for this harmonization. Drug authorities then reached out to the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) to decide about an international, joint industry-regulatory body initiative.
In April 1990, a meeting was held in Brussels, resulting in the birth of the International Conference on Harmonization (ICH). Representatives of industry associations and regulatory agencies of Europe, the US, and Japan met to plan an International Conference.
During the first Steering Committee meeting of ICH, the stakeholders reached an agreement about the terms of reference. They decided that the three criteria on which approval and authorization for new medicines would be given would be Quality, Safety, and Efficacy. It was agreed that harmonization would focus on topics falling under these three critical criteria.
Over the years after its inception, ICH has evolved and grown to make greater harmonization possible to ensure the development of effective, safe, high-quality medicines and their easy registration.
The ICH also developed the concept of Common Technical Document (CTD) and Medical Dictionary for Regulatory Activities (MedDRA).
One of the biggest successes of ICH was the introduction of the concept of Quality by Design (QbD) to the pharmaceutical industry in 2009.
Organizational changes were made in October 2015 and now, ICH comprises 16 Members and 32 Observers.
Founding Members of ICH:
Member countries: United States, Japan, and European Union
Regulatory Representatives:
1. European Commission (EC) and European Medicines Agency (EMA).
2. United States Food and Drug Administration (USFDA).
3. Japan’s Ministry of Health, Labour and Welfare (MHLW).
Industrial Representatives:
1. EU’s European Federation of Pharmaceutical Industries and Associations (EFPIA).
2. USA’s Pharmaceutical Research and Manufacturers of America (PhRMA).
3. Japan’s Pharmaceutical Manufacturers Association (JPMA).
Process of Harmonization
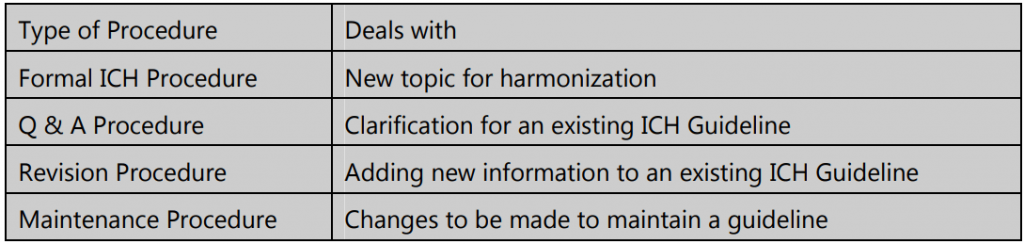
The harmonization activities of ICH may fall into one of four categories: Formal ICH Procedure, Q & A Procedure, Revision Procedure, and Maintenance Procedure.
ICH Procedures
First, a Concept Paper is prepared for the activity to be harmonized. This is a summary of the concept being proposed. Sometimes, a business plan may also be prepared to highlight the cost: benefit ratio of the harmonizing activity.
The formal ICH procedure then begins, in the following steps:
Step 1: Building consensus: Based on the objectives specified in the Concept Paper, a working group prepares a consensus draft called the Technical Document. The working group’s technical experts sign off on this, and the Step 1 Experts Technical Document is submitted to the ICH Assembly with a request for adoption.
Step 2: (a) Based on the report: Assembly confirms that the scientific consensus exists for the technical issues, and the Technical Document may proceed further for regulatory consultation.
(b) This draft guideline is examined and endorsed by regulatory members of the ICH Assembly.
Step 3: This happens in three different stages: Consultation, discussion, and finalization of the Expert Draft Guideline by regulatory members at different levels.
Stage 1: The draft guideline goes to the different ICH regions for discussion in their respective regulatory regions.
Stage 2: All comments obtained during stage 1 are addressed by the expert working group and after discussion, a consensus is reached to prepare the step 3 Experts Draft Guideline.
Stage 3: This draft guideline is finalized and signed by the ICH regulatory member experts. The document is sent to ICH Assembly regulatory members for further proceeding to step 4.
Step 4: ICH Assembly regulatory members agree that sufficient scientific consensus exists on the draft guideline, and it gets adopted as the ICH Harmonized Guideline.
Step 5: ICH Harmonized Guideline is implemented in all the ICH regions through their respective regulatory procedures. Information about when it has become effective is sent to the ICH Assembly and published on the ICH website.
ICH Guidelines: Quality, Safety, Efficacy, Multidisciplinary (QSEM)
The ICH guidelines are covered under four headings under the acronym QSEM – Quality, Safety, Efficacy, and Multidisciplinary.
(a) Quality guidelines: These guidelines cover the areas of quality of drug products such as impurity testing and stability studies and a flexible approach to quality based on GMP risk management.
(b) Safety guidelines: They help to detect potential risks such as genotoxicity, carcinogenicity, and nephrotoxicity. For example, the ICH came up with a non-clinical test methodology to evaluate QT interval prolongation which is probably the most significant reason why drugs have been withdrawn in recent times.
(c) Efficacy guidelines: These guidelines guide designing, conducting, safety aspects, and reporting of clinical trials for pharmaceutical products. Novel drug products derived from biotechnology and genomic/pharmacogenetic techniques for targeted drug delivery are also covered.
(d) Multidisciplinary guidelines: Topics in the pharmaceutical field that do not fit into any of the above categories are covered under this area. This guideline also includes details of (MedDRA), CTD, and standards such as Electronic Standards for the Transfer of Regulatory Information (ESTRI)
Quality Guidelines
Out of all these guidelines, the one most relevant to us is the Quality guidelines. The areas covered under this are labeled from Q1 to Q11 and deal with different aspects of Quality Assurance (QA) relating to pharmaceuticals. Stability testing, analytical validation, impurities, quality systems, risk management, and GMP are some of the most important areas covered.
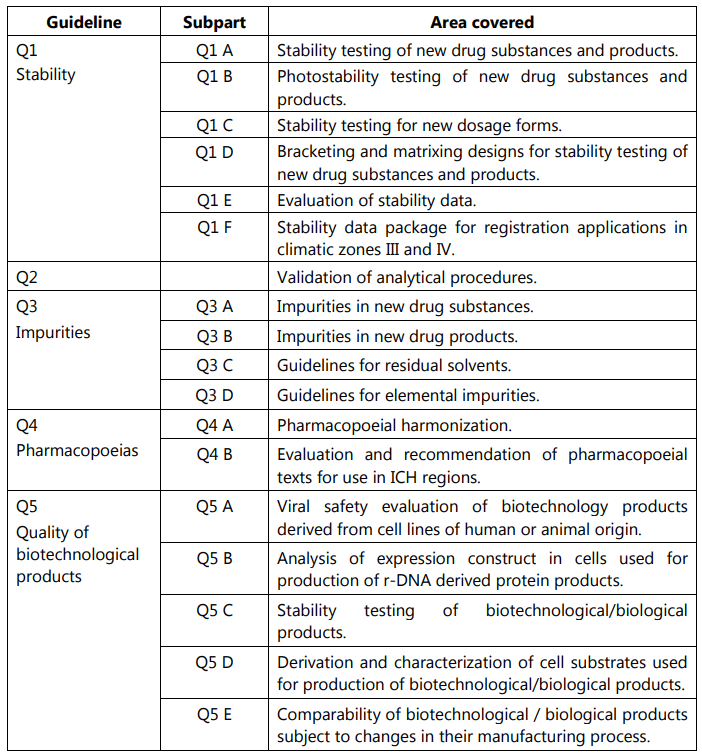
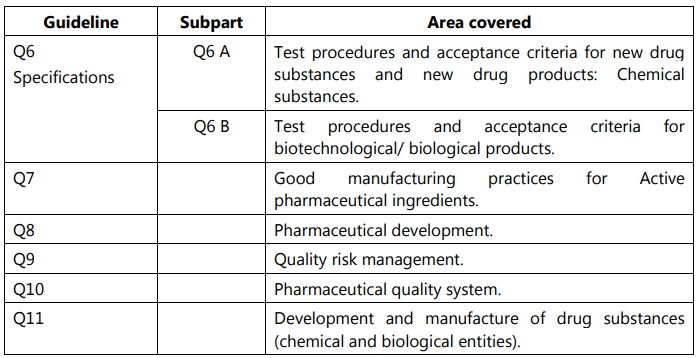
ICH Guidelines for Stability
The ICH guidelines for stability testing define what information must be provided at the time of applying to register a new drug molecule. These guidelines were first adopted in 1993. After revision and updation, the current version in use called Q1A(R2) has been adopted since 2003. This guideline harmonizes the drug registration process for all drugs in the USA, Japan, and the EU. This means a drug registered in one of these regions will not require repeated stability testing when to be sold in any of the other two regions.
Stability testing is important because drug products must be stable when administered to the patients. If an unstable product degrades into toxic metabolites, or if the activity of the drug reduces below 85% of the label claim, there can be serious therapy failures that may even result in death. Stability testing also provides data to choose the formulation parameters, excipients, and the right container-closure system to ensure safe and effective quality products that retain activity throughout the shelf life.
The stability testing data must provide information about how the drug molecule changes over time under different storage conditions. This gives insight into how light, heat, and humidity will influence the chemical nature of the product. Unstable drugs will need specific storage conditions if they have to remain effective. Therefore, it is vital to perform stress testing to study and document the conditions that lead to the degradation of the drug molecule. This information is used to arrive at the shelf life of the drug and what conditions will be optimal for the storage of the product.
Types of Stability Testing
1. Real-time testing: This involves testing drug products for a longer duration to find out what is the maximum product degradation when stored as recommended.
2. Accelerated stability testing: Here, the product is subjected to stress in the form of higher temperatures, moisture, agitation, light, pH, and packaging conditions to study its degradation profile.
3. Retained sample stability testing: This is the testing of samples retained from each batch that has been sent into the market.
4. Cyclic temperature stress testing: Not routinely used. It involves subjecting the products to temperature stresses in a way to mimic likely market storage conditions.
Stability Testing Protocol
This is the written document that describes all major requirements of a well-controlled stability study for a given drug substance or drug product. The basic information to be included in a stability test protocol includes:
- Batch selection – how many batches are to be tested
- Containers and closures that must be used for the testing
- Different positions in which product containers must be kept during testing
- Frequency of drawing samples for analysis
- Overall sampling plan – when and how much to sample and from where
- Test storage conditions based on climatic zone where the drug will be used
- Parameters to be tested to evaluate product stability – mainly the ones expected to change after storage
- Methods to be used for testing, and their validation
- Acceptance criteria for result values, and degradation products
The data obtained by performing the stability studies are used for expiration dating of the drug product and to determine its shelf life.
Overview of ICH Stability Guidelines Contents
Some of the areas covered by the ICH guidelines on stability testing include:
- Stress testing: Study of degradation pathways, effects of change in temperature, relative humidity, pH changes, susceptibility to being degraded by moisture (hydrolysis).
- Photostability testing: Study of the effect of light on drug chemistry.
- Batch selection for stability testing: Not less than 3 primary batches of the drug substance.
- Testing of container closure system: At least thrice; once in 3 months in the first year, once in 6 months during the second year, and then annually.
- Storage conditions for the drug substance and product.
- Storage instructions for different regions and climatic zones, and labeling requirements regarding storage region-wise.
Thanks to the harmonization process of the International Conference on Harmonization (ICH), there are now more than 50 harmonized guidelines. This had led to streamlining of the research and development process and in turn, made it easier to develop and market new medicines to patients all over the globe. There are certainly concerns that non-ICH members are not consulted in the decision-making; however, the educational material provided by ICH is largely beneficial for such countries to streamline their R&D and drug manufacturing efforts.
The membership of ICH has grown over the years of its inception. For ICH to continue to grow and stay relevant, it is necessary to have greater participation from more countries across the world. Plans must consider the involvement of non-ICH members, recognizing and finding ways to overcome the challenges that developing nations face in using the ICH guidelines.
Make sure you also check our other amazing Article on : Total Quality Management (TQM)