WHO Guidelines For Technology Transfer (TT)
Table of Contents
what is a technology transfer?
Technology transfer (TT) is a structural guideline that is intended for the quality of the process, products, standardization, and cost-effective productions.
This is a systematic process in which knowledge and experience are gathered and documented during the life cycle of products originated from development, manufacturing, production, and marketing or commercialization and are transferred to an authorized and accountable organization. Technology Transfer is a fundamental part of the discovery and development of newer pharmaceutical products and dosage forms.
As per World Health Organization, technology transfer is defined as, “A logical procedure that controls the transfer of any process together with its documentation and professional expertise between development and manufacture or between manufacture sites.”
In the Pharmaceutical industry, Technology Transfer is involved in drug discovery, product development, clinical trials, and full-scale commercialization.
Different Terminology Of Technology Transfer (TT)
1.Active Pharmaceutical Ingredients (API): Any ingredients or substances which are used in the manufacturing of a pharmaceutical formulation and are considered as active ingredient of that dosage forms, are called as Active Pharmaceutical Ingredients (API).
2. Change Control (CC): Change control can be defined as, “A formal system by which qualified representatives of appropriate disciplines review proposed or actual changes that might affect the validated status of facilities, systems, equipment or processes. The intent is to determine the need for action that would ensure and document that the system is maintained in a validated state.” (EU GMP Guidelines, Annexure 15).
3. Control Strategy: The proper sets of control obtained from product and process understanding to assure the process performance and product quality which include parameters of products, materials, drug substances, facilities, equipment availability, standardization process, in-process control, quality of finished goods, etc.
4. Critical Control Point (CCP): Some controls are mandatory in the pharmaceutical industry to eliminate or to reduce the quality hazards. CCP is a step at which this control can be applied.
5. Corrective Actions (CA): Corrective actions are taken at CCP while controlling the quality hazards.
6. Quality Assurance (QA): According to WHO “Quality Assurance” is a wide-ranging concept covering all matters that individually or collectively influence the quality of a product. It is the totality of the arrangements made with the object of ensuring that pharmaceutical products are of the quality required for their intended use. Quality assurance, therefore, incorporates GMP and other factors, including those outside the scope of this guide such as product design and development.
7. Quality Control (QC): QC is that part of GMP concerned with sampling, specification, testing, documentation, and release procedures which ensure that the necessary and relevant tests are performed, and the product is released for use only after ascertaining its quality.
8. Design Qualification (DQ): DQ is a documented verification of the proposed design of the facilities, systems, and equipment that are suitable for the intended purpose.
9. Installation Qualification (IQ): IQ is evidence of all key aspects of the process equipment and ancillary system installation adhere to the manufacturer’s approved specification.
10. Operational Qualification (OQ): OQ is established by an objective evidence process for the control limits and action levels in the product of all predetermined requirements.
11. Performance Qualification (PQ): PQ is established by verifying a process, under anticipated conditions, consistently produces a product that meets all predetermined requirements.
12. Drug Master File (DMF): It is detailed information of a specific facility, process, or product that has been submitted to the Medicines Regulatory Authority (MRA) for the incorporation into the application for the marketing authorization.
13. Finished Pharmaceutical Product (FPP): A finished pharmaceutical product will be considered as a product that contains one or more APIs and has undergone all steps of production, standardization, packaging, storing, and labeling.
14. Technology Transfer: Inter-Company Transfer: The transfer of technology between sites of different companies is called intercompany transfer.
15. Technology Transfer: Intra-Company Transfer: The transfer of technology between sites of the same group of companies is called intracompany transfer.
16. Standard Operating Procedure (SOP): It is an authorized written procedure with detailed instructions for the operation of equipment, maintenance of equipment, cleaning of equipment, validation of equipment, environmental control, sampling, and analytical procedures.
17. Technology Transfer Report (TTR): A documented report of technology transfer consists of:
- Procedures
- Acceptance criteria
- Obtained results
- Conclusions
- Deviations, if any
18. Sending Unit (SU) and Receiving Unit (RU): The discipline of any organization involved in transferring of designated process or method is called a sending unit (SU) and the organization involved in receiving the same is mentioned as receiving unit (RU).
General Principles of Technology Transfer (TT)
The basic requirements of Technology Transfer are:
- Quality Risk Management (QRM)
- Documented approach
- Logical approach
- Skilled and trained staff
- Sending Unit (SU)
- Receiving Unit (RU)
The following general principles are to be followed for the successful Technology Transfer:
- The project should attain the quality parameters based on QRM.
- The facilities and equipment available in SU and RU should be similar.
- The trained and skilled staff should be available at RU.
- RU should reproduce the documented evidence of transferred product, process, or manufacturing method against the predetermined specifications of SU.
- Reporting of specifications results and errors by the RU to SU.
- The clarity of the transfer process should be maintained.
- The legal implications like royalties, intellectual property rights, conflict of interests should be conveyed before and during the transfer.
Technology Transfer Protocols
The transfer process should be managed by SU, RU and if required, an additional agency in which proper directions and approvals are provided. There should be a proper management plan and formal agreements for the successful technology transfer.
The following steps should be followed as per the transfer protocol.
- Purpose and objective of the transfer.
- Scope of the transfer.
- Skilled personnel and their responsibilities.
- Comparison of materials, equipments, and methods between SU and RU.
- Documented evidence of each stage of process control and critical stages.
- The transfer of documents should be achieved satisfactorily.
- Assessment of CCP (critical control points).
- Assessment of experimental process for manufacturing.
- Experimental process assessment for standardization and analysis.
- Information of different batches.
- Process validation.
- Assessment of out of specification or deviated results and change control.
- Analysis of finished products.
- Documented reports of analysis.
- Retention of reference materials, active ingredients, intermediate products, and finished products.
- Approval of competent authorities or project manager.
Quality Risk Management (QRM)
Introduction
In the life-cycle of any pharmaceutical product, the quality aspect is very important. The risk of quality variation in the pharmaceutical product can be assessed, controlled, and communicated by a systematic process called QRM. It has been mentioned in ICH guideline Q9.
Principle
The basic principle of QRM is the assessment and evaluation of the associated risks based on scientific knowledge and evidence to maintain the quality of the product and customer satisfaction.
QRM Process
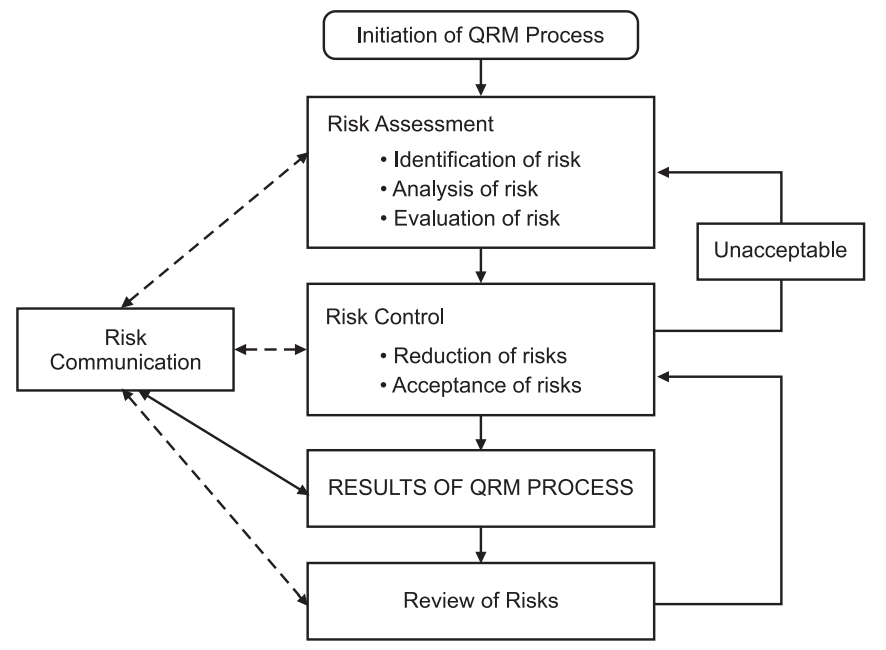
The process of QRM can be summarized in the following different steps (Fig.1):
Step (1) QRM initiation:
To initiate the process of QRM the following plan can be followed:
- Types of risk and problem.
- Questions regarding risks.
- Information regarding quality and potential hazards.
- Information of background and raw data.
- Assessment of required resources.
- Specification of the time limit.
Step (2) Risk Assessment:
This may include the following:
- Identification of Hazards: Systematic process to identify the risks and hazards.
- Analysis of Risks: Qualitative and quantitative estimations of hazards.
- Evaluation of Risks: Comparison of identified and analyzed hazards.
Step (3) Risk Control:
The basic purpose of risk control is to reduce or eliminate the risks. It should be based on the following points:
- The acceptance level of risks.
- Possible steps to reduce or eliminate the risks.
- Balance among benefits, risks, and resources.
(a) Reduction of Risks: The level of risk when exceeds the acceptance criteria, the reduction of risks should be followed. Detection of risk assessment and process control can reduce the level of risks.
(b) Acceptance of Risks: It is the decision to accept the risk.
Step (4) Results of QRM and Risk Reviews:
The review of a result obtained from the QRM process is a part of the quality management system. Results of the QRM process should be documented, reviewed, inspected, audited, and possible change control is suggested. The unsatisfactory review process will suggest the failure of the investigation and the process can be started from the risk assessment step. Step
(5) Risk Communication:
This is the communication process of the data of the risk management system. The risk management system can be communicated at any step of the risk management process (Risk assessment, control, and review process, in Fig.1 mentioned as a dotted line). The well-documented result of the QRM process should be submitted and communicated (in Fig.1 mentioned as a solid line) to the parties.
This guideline is very important in the pharmaceutical industry for QRM. The process is applicable for the pharmaceutical product from manufacturing to the inspection process.
Information Required for Technology Transfer
For the technology transfer from the research and development section to production, the RU should be capable of performing and accommodating the production capacity. The detailed process development should be transferred. At RU, expert personnel and facility available at the site are the primary considerations. Development of protocol by SU and RU jointly for the technology transfer is necessary.
Starting Materials
For the process of successful technology transfer to the production, the specifications and characteristics of starting materials like API and excipients should be identical at both the place SU and RU.
Active Pharmaceutical Ingredients (API)
The complete API master file, Drug Master File (DMF), and relevant auxiliary information of API which are important for the manufacturing process should be provided by the SU to RU. Some important information is mentioned below:
- Details of API manufacturer and supplier.
- Detail scheme of synthesis, process outline, raw materials, process control.
- Details of intermediate products.
- Complete information of API for formulation process. It includes:
– Physicochemical parameters like solubility, partition coefficient (method of determination).
– Particle size distribution.
– Bulk and tap density with a detailed method of evaluation.
– Disintegration profile.
– Nature of hygroscopicity.
– Water content.
– Loss of drying.
– Limit of impurities.
- Microbiological factors.
- Environmental factors.
- Pharmacopoeial standards with a method of determination.
- Stability studies.
- Storage and handling guidance mentioned in Pharmacopoeias.
Excipients
The excipients used in the process of manufacturing have an important role in the quality of the finished product. SU must provide detailed information of excipients to RU. The following information is some of the examples of detailed information:
- Details of manufacturer and supplier.
- Category of excipients.
- Dosage form available.
- Descriptions.
- Solubility.
For transdermal dosage form:
(a) Lipophilicity, Partition coefficient
(b) Particle size and distribution
(c) Specific gravity
(d) Water content and loss of drying
(e) Dissolution rate with detailed process
For solid dosage form:
(a) Bulk and tap density profile with a detailed method of evaluation
(b) Compaction properties
(c) Particle size and distribution
(d) Water content and loss of drying
(e) Nature of hygroscopicity
For semi-solid dosage form:
(a) Melting point
(b) Range of pH
(c) Viscosity
(d) Specific gravity
For liquid dosage form:
(a) Range of pH
(b) Viscosity
(c) Specific gravity
(d) Water content
For parenteral formulation:
(a) Range of pH
(b) Viscosity
(c) Specific gravity
(d) Water content
(e) Osmotic pressure
(f) Ionic strength
For aerosol/inhaled dosage form:
(a) Solubility
(b) Bulk and tap density
(c) Particle size and distribution
(d) Surface area
(e) Water content
Process Information
Regarding the information of process and testing the following information should be provided by the SU.
(a) Requirement of the facility.
(b) Requirement of equipment.
(c) Requirement of the skilled person.
(d) Detail information of raw materials.
(e) Storage guidelines and handling of raw materials and finished goods.
(f) Availability of all SOPs.
(g) Manufacturing process.
- Process flow charts
- Process optimization
- Detail master batch records
- Method of addition of raw materials and excipients
- Details of intermediate products
- Reaction conditions
- Environmental factors
(h) Analytical methods
- Standardization process
- Assay procedure
- Finished goods testing
(i)Validation protocols
- Process validation
- Equipment validation
(j) Annual audits and reviews
(k) Change control, critical control point, and corrective actions
(l) Quality control and assurance
Finished Products
A finished pharmaceutical product is a final product that has completed all stages of production and manufacturing. The finished product should be stored in a specific container and proper labeling is mandatory. All the associated information in a well-documented manner should be informed and transferred from SU to RU. The finished product storage and handling guidelines are to be informed to RU along with the detailed specification and analytical test procedures. The predetermined specifications should be analyzed and the detailed standardization process should be transferred.
Packaging
Information regarding the packaging of the finished product should be transferred to RU. Some of the important instructions are given below:
- Suitable container
- Proper closure system
- Packing material
- Process of packaging
- Design of packaging
- Proper labeling
- Relevant information mentioned in package and label
The information provided by SU should be analyzed at RU for packaging either the packaging is suitable, safe, protective, and compatible with the finished product or not. Packaging should be suggested in such a manner that the final product should not decompose or be affected by environmental factors. The product should not be oxidized and should be protected from sunlight. The formation of undesired substances can make the product spurious and toxic. The container should not react with the product and the efficacy of the product should not be altered by any means after packaging.
Documentation
Some of the important documents required in technology transfer are:
- Technology transfer protocol, qualification protocol, and report.
- Training protocols and reports.
- Standard Operating Procedure (SOP).
- Technology transfer report.
- Analytical methods transfer protocol.
- Validation report (VR).
- Process validation report.
- Cleaning validation protocol and report.
- Validation Master Plan (VMP).
- Masterbatch record.
For a successful technology transfer, the documents related to the facility available at RU, detailed description of the manufacturing process, sampling procedures, approved SOPs for all instruments and process, information of storage, packaging, cleaning, validations, stability information, and regulatory requirements should be provided by SU to RU before starting the productions.
Premises
The SU should make available the information regarding the layout and construction of buildings and services. The air-conditioning system, ventilation, temperature, humidity, and compressed air-related information should be provided to RU before the production. RU should include the risk management, safety requirements, emergency protocols, and waste management provisions in the list of information.
Equipments
The SU should provide the following to RU regarding equipments:
- List of equipments required.
- Specific model and makers of equipments.
- Manuals and SOPs.
- Set-up, maintenance, calibration, and storage protocol.
- IQ, OQ, and PQ status.
Qualification and Validation
The qualification and validation protocol should be decided based on QRM and should be provided by SU to RU in a well-documented manner.
Analytical Method Transfer
Analytical methods are used to analyze raw materials, finished products, packaging materials, and cleaning samples. Analytical method transfer should be performed by providing all the information regarding analytical testing.
The SU should provide the following information for analytical method transfer:
- The methods of analysis and testing of raw materials, finished products.
- Training for analysts and staff.
- Details of equipments used for the testing.
- Testing parameters.
- Experimental principle, design, and methods.
- Quality control testing results.
- Validation reports.
After getting all information from the SU, the RU should have some responsibilities for the successful analytical transfer, some of them are:
- Agreement in acceptance criteria.
- Review of analytical methods.
- Trained and skilled staff.
- Availability of necessary equipments.
- Documents for recording the analytical results.
- Execution of transfer protocol.
- Proper validation to implement the process.
- Availability of Pharmacopoeias.
WHO has mentioned the possible experimental designs for analytical testing. The tests are:
- Identification test.
- Content uniformity.
- Solubility.
- Assay or percentage purity of the components.
- Dissolution parameter.
- Qualitative and quantitative tests for microbiological assays.
- Limit test for impurities.
- Residues recovery.
The responsibilities from both SU and RU should be performed and should prepare the report jointly to execute the transfer protocol.
Agencies For Technology Transfer In India
For the successful TT in India, several agencies are working. Some of them are discussed below.
Asian and Pacific Centre for Transfer of Technology (APCTT)
APCTT is a United Nations Regional Institution that is governed by a Governing Council consisting of a representative designated by the Government of India. The agency is under the Economic and Social Commission for Asia and the Pacific (ESCAP). APCTT was established in 1977 in Bangalore. The main center was moved to New Delhi in 1993.
APCTT governs TT to and from small and medium-scale enterprises in Asia and the Pacific. It regulates the development projects which are funded internationally to provide more strength for TT in Asia and the Pacific.
Technology transfer-related areas of APCTT are institution building, human resources development, studies, business partnership development.
National Research Development Corporation (NRDC)
NRDC was established in 1953 by Govt. of India with the aim of promotion, development, and commercialization of TT from the public sector to the private sector. NRDC is involved in the transfer of technologies, inventions, patents, and processes from the national research and development institutions and universities that are under the administrative control of the Department of Scientific and Industrial Research and the Ministry of Science and Technology.
Technology Information, Forecasting & Assessment Council (TIFAC)
TIFAC, an autonomous organization, was established in 1988 under DST (Department of Science & Technology, Govt. of India). TIFAC is aimed to promote and support technology, innovations in selected areas of national importance. TIFAC concentrates on technology innovation and development through various sustained programs between industry and academia. TIFAC released its Vision 2020 under the leadership of Dr. APJ Abdul Kalam, the former chairman of TIFAC in 16 technology areas, and in 2016 Vision 2035 prepared by TIFAC has been inaugurated by Hon’ble Prime Minister of India Shri. Narendra Modi in 12 thematic areas of national priorities and importance in Mysuru, Karnataka. The 12 thematic areas are:
1. Education
2. Medical Science and Health Care
3. Food and Agriculture
4. Water
5. Energy
6. Environment
7. Habitat
8. Transportation
9. Infrastructure
10. Manufacturing
11. Materials
12. Information and Communication Technologies (ICT).
Biotech Consortium India Limited (BCIL)
BCIL, a public limited company, was inaugurated in 1990 under the Indian Companies Act, 1956. BCIL is promoted by the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India, and All India Financial Institutions. BCIL was aimed at providing the necessary linkages among stakeholders and business support to facilitate the acceleration of the commercialization of biotechnology. BCIL assists scientists, technologies, research institutions, universities, first entrepreneurs, the corporate sector, national and international organizations, central government, various state governments, banks, and financial institutions.
BCIL works in the following aspects:
• Technology transfer
• Project consultancy
• Fund syndication
• Information dissemination
• Manpower training and placement related to biotechnology
Technology Bureau for Small Enterprises (TBSE)
TBSE provides the opportunity to the small enterprises at the global level for the acquisition of technology or establishes business collaboration. TBSE works under Development Commissioner, Ministry of Micro, Small and Medium Enterprises (MSME) and it is partially funded by the office of Development Commissioner (DC), Small Scale Industries (SSI), and Government of India.
The important features of TBSE are:
1. TBSE offers a professionally managed system for technology and collaboration search.
2. TBSE builds up confidence between partners.
3. TBSE has a proper mechanism for arranging technology and finance.
4. TBSE provides a gateway to the global technology market through networking.
5. TBSE takes up project appraisal and preparation o the business plan.
Small Industrial Development Bank of India (SIDBI)
SIDBI was established on April 2, 1990, through an Act of Parliament, under the Department of Financial Services, Government of India. It is a development financial institution in India. The headquarter is situated in Lucknow, Uttar Pradesh. The purpose of SIBI is to provide refinance facilities and short-term lending to industries and serves as the principal financial institution in the Micro, Small, and Medium Enterprises (MSME) sector.
Technology Transfer Related Documents
Confidentiality Agreement
It is also called a non-disclosure agreement (NDA). It is used to protect the proprietary nature of the technology and retain the confidentiality of technology or invention. The drafting of the appropriate clauses can be essential for the maintenance of the value of the technology. The need for this agreement is due to the increase in competition and the new technologies can be exploited. Thus it is necessary to obtain protection to the continuous innovation process through confidentiality agreements.
Licensing
The license agreement is generally referred to the licensing of intellectual property rights such as; patents, trademarks, copyrights, etc. This agreement has a role in maintaining the confidentiality and secrecy aspects of the contract.
MoUs
MoU stands for Memorandum of Understanding. It is a negotiated agreement and contract between the Government and the Management of the Central Public Sector Enterprise (CPSE). MoUs are used either when the parties do not imply a legal commitment or where the parties cannot create a legally enforceable agreement.
Make sure you also check our other amazing Article on : Pilot Plant Scale-Up Techniques