Just as measuring instruments need to be calibrated, it is important to demonstrate that the equipment and utilities in a pharmaceutical company are appropriate for their intended use, and to prove that they are performing as expected. The activities undertaken to generate such evidence are called Qualification.
Definition
Table of Contents
Qualification may be defined as the act of proving and documenting that given equipment or process or utility is correctly installed, working properly, and is consistently producing the expected results.
Qualification is a part of the process of validation; however, qualification alone does not complete process validation.

Phases of Qualification
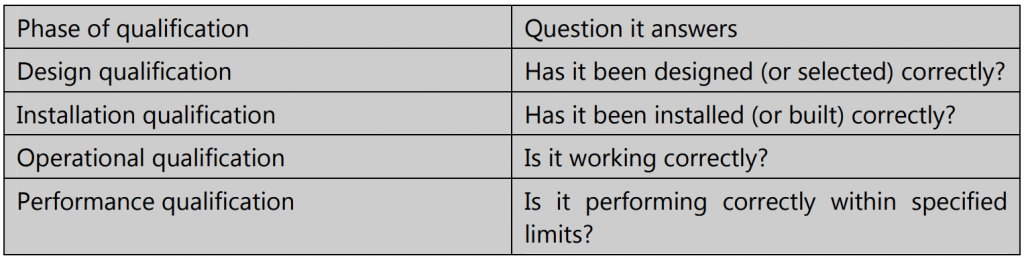
There are four major phases of qualification – Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ).
(a) Design Qualification is defined as documented verification that the proposed design of equipment, systems, and facilities is appropriate for the intended purpose. DQ must be performed whenever purchasing new equipment; it should also be performed when existing equipment is going to be used for any new application.
(b) Installation Qualification is defined as documented evidence that the equipment, supporting utilities, and premises have been built or installed in keeping with design specifications. IQ serves to verify that equipment installation has been done as recommended by the manufacturer, correctly, and kept in a suitable environment.
(c) Operational Qualification is defined as establishing documented evidence that any equipment, facility, or utility functions as intended, in keeping with its operational specifications. OQ thus helps to verify that the installed equipment works correctly.
(d) Performance Qualification is defined as establishing documented evidence that the process works to consistently produce a product that meets all the predetermined quality requirements. PQ helps to verify that performance within the specified limits is as expected.
WHO Definitions
DQ – Documented evidence that the premises, supporting systems, utilities, equipment, and processes have been designed by the requirements of good manufacturing practices.
IQ – The performance of tests to ensure that the installations (such as machines, measuring devices, utilities, and manufacturing areas) used in a manufacturing process are appropriately selected and correctly installed and operate by established specifications.
OQ – Documented verification that the system or subsystem performs as intended overall anticipated operating ranges.
PQ – Documented verification that the equipment or system operates consistently and gives reproducibility within defined specifications and parameters for prolonged periods.
Scope of Qualification Phases
Considerations While Doing Qualification
The following factors must be considered when doing qualification:
1. Qualification must be performed as per predetermined and approved qualification protocols.
2. Results must be recorded and should be a part of the qualification reports.
3. Areas or rooms must be qualified before utilities; utilities must be qualified before equipment.
4. Equipment can be used only after it has been qualified and documented evidence shows that it is suitable for the intended purpose.
5. Qualification should be done in a logical sequence – first DQ, then IQ, then OQ, and finally PQ.
6. Some stages of qualification may be performed by a third party; however, the final responsibility of ensuring the qualification is done as per GMP rests with the contract-giver.
7. All documents related to the qualification process (specifications, standard operating procedures, manuals, acceptance criteria, and certificates) should be maintained.
8. All equipment, utilities, and systems must be maintained in a qualified state at all times. When necessary, they must undergo periodic requalification.
9. Validation of processes must be done using qualified equipment only.
Requalification
When any equipment undergoes any kind of modification or relocation on a scale that has a direct impact on the product quality, it must undergo a re-qualification. This process must be handled with a documented change control procedure, after due review and authorization.
Factory Acceptance Test and Site Acceptance Test
In some situations, equipment, or a system or utility may be assembled fully or partially at some other site. In such cases, testing and verification must be performed to ensure it is fit for dispatch, and results must be recorded in the Factory Acceptance Test report before shipment of the item.
After the shipment is received by the end-user, tests must be repeated to verify that the equipment or system, or utility is of acceptable quality. Results of these tests must be recorded in the Site Acceptance Test report.
Make sure you also check our other amazing Article on : Quality Audit