Good Documentation Practices: Documents must be designed and prepared with care. They must be accurate and written in a way that ensures consistency and prevents any errors.
- Content in the documents must be unambiguous. All documents must have a title, nature, and purpose that must be clearly stated. Content must be arranged in an orderly manner and allow for easy checking.
- Full-text spellings must be used the first time, with the abbreviation in brackets. Subsequently, abbreviations may be used in the rest of the document.
- All documents must carry an effective date, and review date if applicable.
- All documents must be approved; they must bear the signature (with date) of the persons preparing, reviewing, and approving the document.
- All documents must have a distinct identification number and version number.
- Documents must be reviewed periodically and in case of revision, the version number must change. Only the most current version must be available; systems must exist to prevent the use of documents that have been superseded.
- No document must be handwritten. However, there may be a provision for the entry of data by hand (for example, in the Batch Manufacturing Record, where real-time data entries are made). Sufficient space must be available for making the entries, which must be legibly made with indelible ink. Using temporary recording on scraps of paper etc. is prohibited.
- If a correction is to be made to a record/entry, it must be made in such a way that the original information is not defaced. Overwriting and use of white ink or correction fluid or sticky notes to make corrections is prohibited; however, it is acceptable to strike through the incorrect entry with a single line and write the correct entry next to it. The correction must be accompanied by a signature/initials and dated. Where necessary, the reason why the correction has been made must also be recorded such as ‘typographical error’ or ‘calculation error’ or ‘recording error’.
- Entries in the record must be made immediately – that is, at the time of the particular action.
- Signature and date must accompany the entry where specified. No person should sign for another staff unless specifically authorized. Forging of initials or signatures is prohibited.
- Master copies of documents must be stored securely. Only authorized persons must have access to these. Copies may be made for issuing; while handing over the copies, details have to be recorded of who issued the document, to whom, and the date and time of issue.
- Copies reproduced from master documents must be clear enough to allow comprehension.
- If printouts are obtained on thermal paper from an instrument, they must be photocopied (as the print will fade over time). Both the original and photocopy labeled as “Copy of original” must be attached to the report.
- Critical records must be stored securely, protected against loss, falsification, or any damage by the elements, with limited access to only authorized persons.
- Copies of critical records must be available in electronic form or paper or microfilm and be stored separately from the originals.
- In the case of electronic data processing methods, access to the master templates and documents must be given only to authorized persons to enter data or modify it in the computer. Passwords and biometrics may be used to control access.
- If electronic signatures are to be used, they must be unique to the person. Such electronic signature is generally using an identification code that comprises a username and password. Using the saved digital image of the hand-written signature of the person is not permitted.
- Retaining important documents (such as developmental history, technical reports, validation reports, production and control records, training records, distribution records, etc.) must be done in keeping with regulatory requirements. For example, records about the production, testing, and distribution of a batch of drug products must be retained for a minimum of 1 year following the expiry date of that batch.

Common Documentation Errors
1. Illegible entries.
2. Falsified data (date/time/version number).
3. Signatures missing.
4. Raw data to support results missing.
5. Missing documents.
6. Discarding data that does not match requirements.
7. Invalid and fabricated data.
8. No recording of activities.
9. Data manipulation/deletion.
10. Incomplete records.
Excerpt from a warning letter issued by USFDA to a firm citing lack of adequate documentation practices.
“…Your quality unit (QU) lacks appropriate responsibility and control over your drug manufacturing operations.
During the inspection, our investigator observed discarded cGMP documents and evidence of uncontrolled shredding of documents. For example, multiple bags of uncontrolled cGMP documents with color-coding indicating they were from drug production, quality, and laboratory operations were awaiting shredding. Our investigator also found a blue binder containing cGMP records, including batch records for U.S. drug products, discarded with other records in a 55-gallon drum in your scrap yard. cGMP documents in the binder were dated as recently as January 21, 2019: seven days before our inspection. Your QU did not review or check these documents before disposal….”
“…The uncontrolled destruction of cGMP records, and your lack of adequate documentation practices, raise questions about the effectiveness of your QU and the integrity and accuracy of your cGMP records….”
“…Your quality system does not adequately ensure the accuracy and integrity of data to support the safety, effectiveness, and quality of the drugs you manufacture…”
Types of Documents
The records and documents in the pharmaceutical industry are categorized into the following classes:
1. Primary records (contracts, production formulae, packing instructions, supply source documents, etc.).
2. Procedures or supporting documents (SOPs, instructions, manuals, guidebooks).
3. Subsidiary records (equipment/instrument printouts, calibration reading reports, etc.).
4. Quality control records (test methods, test results, investigations, internal audit reports, Corrective and Preventive Action (CAPA) reports, recall files, etc.).
Commonly Used Documents
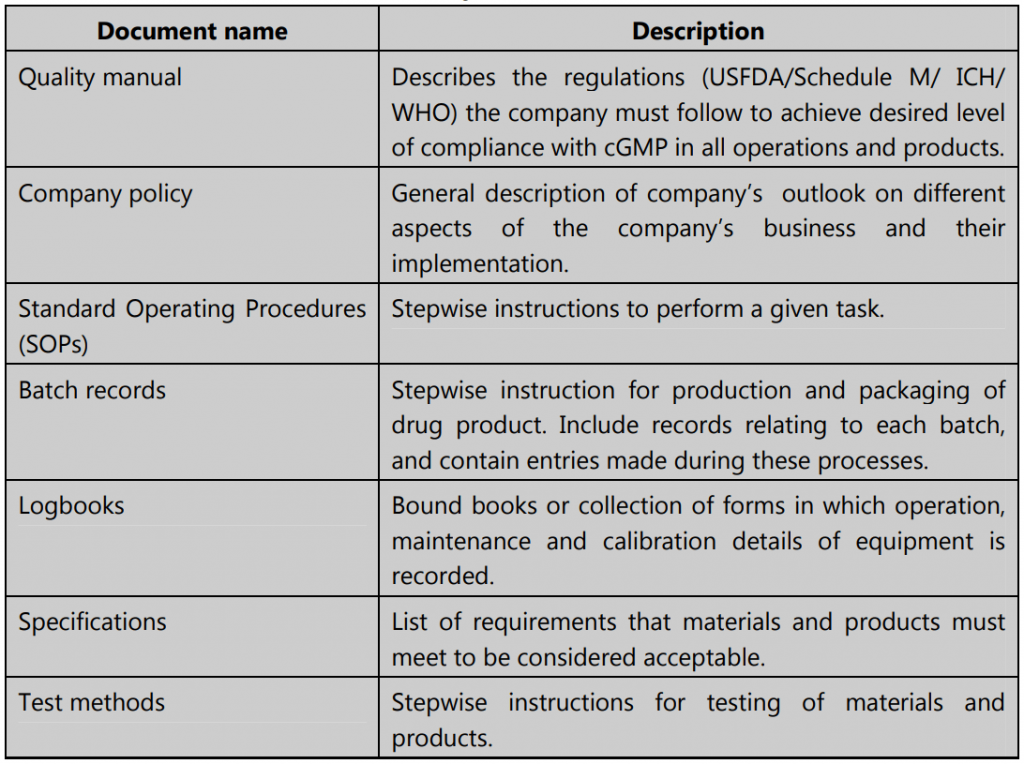
Importance of Documentation
Proper documentation is the backbone of current Good Manufacturing Practices (cGMP) and in the regulatory world, it is commonly held that “If it isn’t documented, it wasn’t done!”
Documentation is the most essential part of quality management and quality assurance system for the following reasons:
- Documents are evidence of all manufacturing and testing activities and provide traceability to verify if certain actions were performed or not.
- Written procedures provide clarity and ensure there are no errors that may arise during spoken communication.
- Records, documents, and reports give a clear picture of what has been done and is ongoing work, and it also helps to plan better for the future.
- A comprehensive review of the documents maintained in a pharmaceutical facility is often the key used by regulatory bodies to assess the quality function of the facility.
- Accurate and clear records allow the critical reviewing of processes, which can help to improve quality and create cost-saving measures too.
- Good documentation is a must to attain ISO certification and any other industry-specific certifications.
Make sure you also check our other amazing Article on : Drug Recall