
Antiviral Drugs: Viruses represent a separate and unique class of infectious agents, that can replicate only inside the living cells of organisms. Outside the host cell, they are like any other nonliving entity and are referred to as virion or virus particles. The smallest viruses possess a diameter of not more than 20 mm while in the case of the large viruses, the diameter may go up to 300 mm. The constitution of a virus is much more simple as compared to that of bacteria. According to Lwoff, an infectious agent that:
- possesses simple chemical composition.
- lacks the metabolic enzyme machinery.
- lacks the protein-synthesizing system.
- contains only one type of nucleic acid (i.e. either DNA or RNA), and
- possesses a host-cell-dependent machinery of multiplication that can be named a virus.
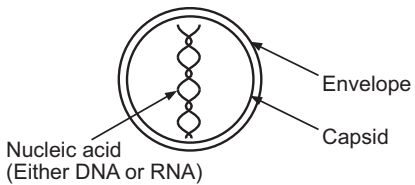
Unlike bacteria, viruses do not possess cell-wall. Viruses consist of one or more strands of linear or helical strands of either DNA or RNA enclosed in a shell of protein known as the ‘capsid’. The capsid is composed of several sub-units known as capsomeres that decide the shape of the capsid. Though often spherical, capsids may possess different shapes. In certain cases, the capsid may be surrounded by an outer protein or lipoprotein envelope. This encircling membrane may be called an ‘envelope’.
Since the biology of viral replication is dependent on the host cell metabolic machinery (e.g. protein synthesis, various enzyme systems), unlike bacteria, viruses can not be grown on the nutrient media. They can replicate only in the host cell which may be a bacteria, animal, or plant cell. Hence, viruses are considered obligatory intracellular parasites that utilize biochemical machinery and many products of the host cell to sustain their viability.
Viral diseases include influenza, smallpox, rabies, poliomyelitis, yellow fever, ornithosis, psittacosis, and lymphogranuloma venereum.
The viral replication may be outlined as under:
1. Adsorption: The virion invades the host cell membrane. The reactive sites on the capsid firmly bind with their complementary sites on the host’s cell. The viral particle is encapsulated by the host’s cell cytoplasm, forming a vacuole.
2. Uncoating: The genetic material or viral genome (DNA or RNA) passes into the host’s cell, leaving the capsid covering outside the host’s cell. Sometimes uncovering of the viral genome occurs within the host cell. This step is referred to as penetration into the host’s cell. Only the viral genome is infectious to the host’s cell and the capsid of the protein coat determines the site of the attacks of the virus within the hosts. The viral genome is different from the host’s nucleic acids and hence it is infectious. Sometimes due to its proteinaceous nature, capsid contents may turn out to be antigenic to the host cell and initiates several immunological reactions to the host.
3. Synthesis of viral components: Viral genome enters the cytoplasm or nucleoplasm and directs or utilizes the host’s nucleic acid machinery for the synthesis of new viral protein and the production of the more viral genome. Thus, it not only consumes the actual material for its use from the host’s cell but also enjoys the services of the biochemical systems of the host to incorporate this material into the several proteinous subunits needed for its replication.
4. Release of the virus: Viral nucleic acid and capsid protein materials are synthesized in different parts of the host cell by the host-cell ribosomes. The m-RNA is synthesized from the viral genome. The host cell machinery, however, fails to differentiate between viral and cell-directed orders. A large number of newly synthesized viral particles then have to be brought together for assembling new virions. The latter is released from the cell by the budding process. They may acquire the lipoprotein envelope at the time of their release. The new virions then invade the fresh host cell and repeat the whole process. Since the host cell machinery is utilized for the production of new virions, the normal cell functions cease at the time of replication. Some viruses induce the production of toxic intermediates that adds to their pathogenicity. Viruses are composed of one or more strands of a linear or helical nucleic acid core, consisting of either DNA or RNA, but not both. Viruses thus can be classified as per the type of nucleic acid present in them. The types are:
(A) DNA viruses: This class includes pox viruses, papovaviruses, adenoviruses, and herpes viruses.
(B) RNA viruses: This class includes, arboviruses, myxoviruses, picornaviruses, and rhinoviruses.
Design of Antiviral Agents: Viral chemotherapy is still in the phase of infancy. Although a great deal of work has been done, it has resulted in the development of only 3-4 clinically used agents. Presently immunization, public health measures, and physical and chemical disinfection procedures play a vital role in the prevention of the spread of viral diseases.
Viruses are essentially intracellular parasites. Unlike bacteria, viral replication is dependent upon the energy, proteins, and enzymatic machinery of the host cell. Bacteria have self-contained biosynthetic machinery. Hence, the drug in chemotherapy enjoys the advantage of selective attack on bacteria due to many metabolic and molecular differences between the pathogen and the host cell. Since viruses take over the metabolic machinery of the infected human cell, a close relationship exists between the multiplying virus and the host cell. Virus replication is intimately dependent upon the host’s cell metabolism. This fact severely limits the usual opportunities to design antiviral agents having a selective effect on the viral cell. Attempts to inhibit viral growth without damaging the host cells became fruitless.
Viral infection or its presence in the host cell is felt so late that it results in extensive viral multiplication and tissue damage. The late diagnosis and recognition of the disease state projects almost negligible chances for effective therapy. The drugs become useless, even if they are made available.
The important key events in the viral replication are diagrammatically shown in Fig.2.
Various steps involved in viral replication could be successfully utilized as the basis for designing antiviral agents. For example, the following sites in viral replication offered promising points for the attack of antiviral agents.
- Adsorption of the virus on the host cell,
- Penetration of virus in a host cell, and
- Synthesis of viral genetic material.
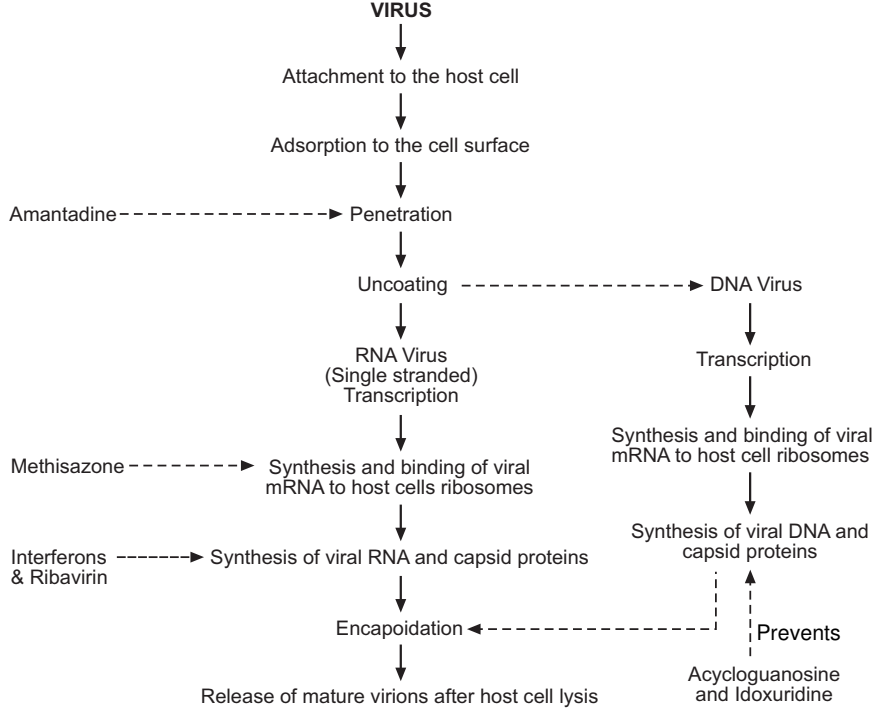
Amantadine hydrochloride inhibits viral penetration and prevents influenza while methisazone and idoxuridine inhibit viral DNA synthesis. Due to one or more reasons (e.g. narrow activity spectra or toxicity) not a single drug in this area, enjoyed clinical popularity. The development of an antiviral agent having selective toxicity to viral cells leaving host cells unaffected remains a dream for medicinal chemists.
Antiviral agents have often been proven to be disappointing due to the following problems in their development:
- Lack of satisfactory experimental models.
- Use of the wrong virus in the laboratory.
- Narrow spectrum of activity.
- Limitations on uses due to their toxicity.
- Difficulties in their clinical assessment.
These disappointing features of antiviral therapy force us to accept vaccines as the best prophylactic agents in the treatment of viral diseases. There are four forms of antigen used in vaccines. These include,
- Attenuated living,
- Killed by the chemicals, such as formalin,
- Toxoids (i.e. toxins which through the application of heat or formalin are converted to non-toxic form).
- Subunits are prepared by purifying important antigens of microorganisms.
Vaccination can be effectively used to prevent measles, rubella, mumps, poliomyelitis, yellow fever, smallpox, and hepatitis B. They have only prophylactic utility and remain ineffective once the infection has occurred and spread within the host.
Classification
Table of Contents
Various antiviral agents so far introduced, are found to possess a very narrow spectrum of activity. For example, agents which are effective against DNA viruses are usually ineffective against RNA viruses, and drugs that show activity against RNA viruses remain silent against DNA viruses. During antiviral therapy, the drug used receives a very fast acknowledgment from the viruses, resulting in the rapid development of drug-resistant strains of viruses.
The currently available antiviral drugs can be chemically classified as:
- Purine nucleosides and nucleotides
- Pyrimidine nucleosides and nucleotides
- Thiosemicarbazones
- Benzimidazoles
- Adamantane amines
- Interferons
- Miscellaneous agents
(1) Purine Nucleosides and Nucleotides: Acyclovir triphosphate (Acycloguanosine)
It is a synthetic purine nucleoside analog in which a linear side-chain (i.e. –CH2OCH2CH3) is attached at the 9th position instead of the cyclic sugar present in the guanosine molecule.
Acyclovir undergoes a phosphorylation process with the help of viral thymidine kinase enzymes to form acyclovir triphosphate. The latter selectively inhibits herpes virus DNA polymerase. The faulty transcription in viral DNA leads to the inhibition of the virus replication process. The affinity of viral thymidine kinase enzymes for acyclovir is about 200 times greater than that of mammalian enzymes for the drug. This explains the selectivity of attacks on viruses.
It can be used orally, topically, and intravenously. About 15% of the administered dose is bound to the plasma proteins. It has a plasma half-life of about 2.5 hours. It is excreted in urine mostly in unchanged form along with a minor amount of inactive metabolite, 9-carboxy methoxy methylguanine.
Adverse effects include nausea, headache, amnesia, hypotension, tremors, and coma. Local irritation, ulceration, and burning may occur when it is applied to genital lesions.
It is effective against herpes virus infections, herpes zoster infections, and infections caused by varicella zoster and cytomegalovirus. It may be used orally for the treatment of both initial and recurrent episodes of genital herpes. A 3% acyclovir ointment may be used for the treatment of ocular herpetic infection and herpes keratoconjunctivitis. It may also be used intravenously in the treatment of herpes simplex encephalitis. However, the development of resistance to acyclovir may limit its clinical utility.
(2) Pyrimidine Nucleosides and Nucleotides
(i) Idoxuridine: It was discovered in 1959 by Prusoff. It is a topically used antiviral agent chemically related to trifluridine and is effective against vaccinia and herpes simplex virus, pseudorabies, B. virus, myxoma virus, polyoma-virus, and some papovaviruses.
Idoxuridine is chemically very similar to thymidine, a compound that in the normal cell undergoes phosphorylation and then gets incorporated into a DNA molecule. Due to the structural similarity with thymidine, with the help of thymidine kinase and thymidine monophosphate kinase enzymes, it is converted to the active triphosphorylated derivative. The latter blocks the DNA polymerase enzyme resulting in interference in nucleic acid and protein synthesis in DNA-viruses. It also prevents the assembling of viral components by inhibiting the synthesis of a protein required for their assembly. Since the host cell DNA synthesis is also affected, it produces host cytotoxicity. Hence, its systemic use is not recommended. It is thus usually given topically. Hence, serious side effects rarely attend to its use.
It is available in the form of ophthalmic ointment (0.5%) and ophthalmic solution (0.1%). It gets rapidly inactivated by deaminases or nucleotidases into 5-iodouracil and inorganic iodides. Adverse effects include pain, irritation, pruritus, inflammation or edema of conjunctiva and eyelids, corneal vascularization, lacrimation, stomatitis, neutropenia, and thrombocytopenia. It is effective in the treatment of herpes simplex infections of the eyelid, conjunctiva, and cornea.
(ii) Trifluridine: It is a halogenated thymidine analog used as a topical antiviral agent. It is effective against herpes simplex virus, vaccinia, and adenovirus. It is converted to the active 5-monophosphate form. Due to the structural similarity with thymidine, it gets incorporated, instead of thymidine phosphorylated form, into replicating viral DNA. This results in the wrong expression of genetic information due to abnormal base pairing. It also blocks thymidylate synthetase and deoxythymidine kinase enzymes in the viruses. The overall biological effects may get reflected into:
- increased number of errors in viral protein synthesis,
- increased rate of mutation, and
- inhibition of the viral replication process.
It has a plasma half-life of 12 – 18 minutes. Metabolism involves the removal of deoxyribose by nucleoside phosphorylase with conversion to trifluorothymine. Urinary excretion of parent and metabolites is rapid. Adverse effects include stomatitis, reticulocytopenia, hypophosphatemia, anemia, neutropenia, megaloblastic, and hypocalcemia.
In the form of a 1% ophthalmic solution, it is used to treat certain ocular infections (e.g. ocular keratitis and keratoconjunctivitis). It exhibits effectiveness in cases where idoxuridine treatment fails.
(iii) Vidarabine (Adenine arabinoside) (Ara – A): Vidarabine is a purine nucleoside. This antiviral agent is isolated from the bacteria, Streptomyces antibioticus. It is an analog of adenosine originally developed for the treatment of leukemia. It is converted in-vivo enzymatically to ara-ATP. The latter impairs early steps in viral DNA synthesis, presumably by inhibiting viral DNA polymerase. It exhibits considerable host toxicity due to part inhibition of cellular DNA polymerase enzymes.
It is effective against vaccinia, herpes simplex virus, cytomegalo virus, varicella zoster, pseudorabies, and myxoma virus. It may be used topically as a 3% ophthalmic ointment or as an I.V. infusion. About 20-30% administered dose is bound to the plasma proteins. It has a plasma half-life of 1.5 hours. In the cornea and the plasma, it metabolizes to arahypoxanthine (active; the half-life is 3.3 hours) by xanthine oxidase enzyme through a process of delamination.
Adverse effects include nausea, diarrhea, thrombophlebitis, hallucinations, tremors, dizziness, burning, itching, and redness of the eyes. It should not be used to treat trivial infections.
It is used to treat herpes simplex infections in neonates. It is also used to treat herpes simplex encephalitis and herpes zoster infections in immuno-compromised patients. It may have activity in cytomegalovirus infections and type B virus hepatitis. In the form of 3%
ophthalmic ointment, it may be used topically to control recurrent epithelial keratitis and kerato-conjunctivitis. However, it remains ineffective against bacterial, fungal, and adenovirus infections. Its antineoplastic potential is under investigation.
Vidarabine-5′-monophosphate and vidarabine–hypoxanthine – 5′ monophosphate are the vidarabine derivatives that are currently under investigation for their utility in antiviral therapy.
(iv) Ribavirin: It is a synthetic nucleoside chemically related to inosine, guanosine, and xanthosine. It is a broad-spectrum antiviral agent effective against nearly all major viruses. It has virustatic properties. It is in-vivo converted to ribavirin – 5′ – monophosphate which acts as a competitive inhibitor of inosine 5′ monophosphate dehydrogenase. This results in the inhibition of guanine monophosphate synthesis, followed by the inhibition of viral RNA synthesis.
It has a plasma half-life of 9 hours. It is extensively metabolized in the liver. Principle metabolites include mono, di, and triphosphate derivatives, tricarboxylic acid analog, and 1, 2, and 4-triazole carboxamide metabolite. Deribosylation and breakdown of triazole ring are also reported to occur.
Adverse effects include rash, conjunctivitis, blurred vision, reticulocytosis, dyspnea, chest discomfort, dizziness, anemia, and hypotension. It is contraindicated during pregnancy.
It is an investigational antiviral agent used in the treatment of infections due to respiratory viruses. It is used in the form of aerosol to treat severe lower respiratory tract infections due to respiratory syncytial virus (RSV). It is also undergoing evaluation in the treatment of human immunodeficiency virus (HIV) infections, such as AIDS.
Adenine arabinoside and cytosine arabinoside are other examples from this category. Cytosine arabinoside (cytarabine) is effective against herpes viruses. It is presently used in cancer chemotherapy.
(3) Thiosemicarbazones
In 1947, Domagk (sulfonamide fame) reported that some derivatives of benzaldehyde thiosemicarbazone protected laboratory animals against tuberculosis. This initiated an extensive investigation of the thiosemicarbazones during which the activity of methisazone (N-methylisatin – β thiosemicarbazone) was discovered. It is effective against a variety of poxviruses and some RNA viruses.
The antiviral action of thiosemicarbazone is perhaps due to the formation of metal chelate with various metal ions including Cu, Zn, Ni, ferrous, and manganese. Methisazone thus acts by interacting with metalloenzymes that are necessary for the replication of certain viruses. The thiosemicarbazones may also react directly and specifically with the viral nucleic acid.
Methisazone prevents the replication of vaccinia viruses. It is poorly absorbed by the oral route. Major metabolic pathways include demethylation, replacement of sulfur by oxygen in the
side chain, and hydroxylation in the aromatic ring. Isatin – 3 – thiosemicarbazone is the active metabolite retaining half the activity of the parent drug.
It is used as a prophylactic agent against small-pox and for the prevention and treatment of generalized vaccinia or vaccinial encephalitis.
(4) Benzimidazoles
The following two compounds of this family are found to show promising results in trials.
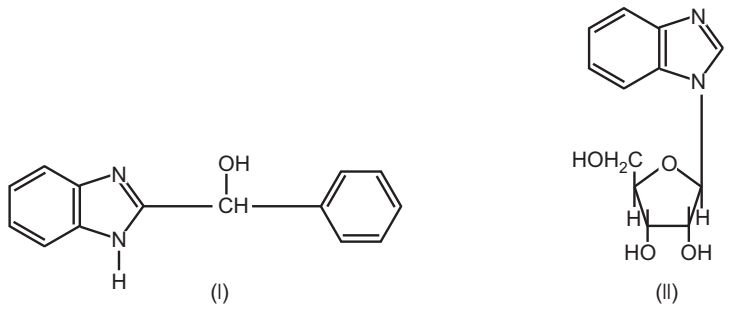
Both these agents do not interfere with viral adsorption, penetration, and uncoating. They appear to act by inhibition of viral RNA synthesis.
(5) Adamantane Amines
Amantadine hydrochloride: It was first reported by Davies et al in 1964. It is a synthetic antiviral agent effective against infections caused by influenza A viruses including H3N2 and H1N1 subtypes. In higher concentrations, it is also effective against influenza B, rubella, and other viruses.
Amantadine allows viral adsorption to the host’s cell but inhibits the uncoating of the influenza virus and prohibits penetration of the viral genome into the host’s cell. Because the virion remains adsorbed to the host’s cell surface, it becomes susceptible to attack by the host’s antibodies.
It is orally active and is completely absorbed from GIT. It has a plasma half-life of about 9 – 37 hours. It is not metabolized in the body and is excreted almost in unchanged form in the urine. The rate of urinary excretion depends on the urinary pH; acid urine facilitates rapid excretion.
Adverse effects include GI upset, nervousness, insomnia, tremors, confusion, hallucination, and coma. It is contraindicated during pregnancy and in patients with mental illness and epilepsy.
Besides its antiviral potential, it may also be employed in the therapy of parkinsonism. Rimantadine is a better-tolerated and less toxic analog of amantadine which is currently undergoing clinical evaluation. It has similar actions and uses.
(6) Interferons (Antiviral Proteins)
The presence of interferons was first reported in 1957 by scientists Issues and Lindermann at the National Institute for Medical Research, U.K. The term is applied to a class of glycoproteins of molecular weights from about 20,000 to 50,000. Each contains approximately 150 amino acids. Interferons are produced from the host’s cell when it is infected or exposed to an inactive virus. The endogenous synthesis of interferons is under the control of host cell RNA. Three major types of human interferons are designated as α, β, and γ according to antigen specifications. The preferred abbreviation of interferon is IFN. Interferons can also be designated as per the source. For example, human (Hu IFN), bovine (Bov. IFN), murine (Mu IFN), etc. Currently, a major source of human interferons is white blood cells that have been cultured and then exposed to appropriate viruses. In the body, after their release, they may attach to surface receptors of the adjacent cells and initiate the production of additional interferons.
Viruses differ in their ability to induce the synthesis and/or liberation of interferons. The adsorption of a virus to the host’s cell or the fully infected condition of the host’s cell induces the formation and release of interferons. This is a sort of immunization process. The release of interferon imparts resistance to the person against the attack of viral infection. Generally, a person once infected with one virus develops resistance against other viral infections. This phenomenon is known as ‘viral interference’. In non-infected cells, it can induce the formation of a second inhibitory protein which can prevent the transcription of any viral m-RNA that might subsequently be produced in that cell.
Interferons are characterized by the following features:
- They are non-toxic to the host cell.
- They exhibit antiviral activity in extremely low concentrations, and
- They do not possess antigenic activity.
Interferons appear very soon and sometimes within seconds after the viral attack. However, they lack specificity. Once released from the infected host cell, interferons induce the synthesis of translational inhibitory proteins in other non-infected cells. These proteins impair the translation of viral m-RNAs and thus interfere in viral replication. The synthesis of viral RNA polymerase and viral thymidine kinase is inhibited. The transcription of the viral genome is also found to be inhibited.
Interferon preparations may contain more than one type of interferon. These exhibit a uniquely broad spectrum of antiviral activity in-vitro and show promising effects when used in combination with vidarabine. Interferon preparations are effective against infections caused by varicella zoster, encephalomyocarditis virus, vesicular stomatitis virus, rabies, vaccinia, influenza B, and cytomegalovirus.
Interferons are not orally absorbed. They are usually given by i.v. infusion and have a plasma half-life of about 15 – 20 minutes. Metabolic pathways for interferons are not still known.
Adverse effects mainly include fever, reticulo-cytopenia, neutropenia, and thrombocytopenia. They are clinically effective against chronic active hepatitis B viral infection, cytomegaloviral infection, congenital rubella infections, and respiratory viral infections. Interferons are also showing promising results in the therapy of some cancers like melanoma, multiple myeloma, osteogenic sarcoma, certain leukemias, and breast cancer. Their use in organ transplantation is also being extensively studied. Interferon synthesis and liberation in animals can be induced by:
1. Biological inducers: These include human leukocytes, fibroblasts or lymphoblastoid cells, and
2. Chemical inducers: These include:
- Polyriboinosinic polyribocytidilic acid is a complex of polyinosinic and polycytidylic acids. Structurally, this complex is a synthetic analog of a double-stranded RNA,
- A new group of 6-phenyl pyrimidine derivatives induces the host cell to produce interferons, and
- Tilorone hydrochloride.
All these interferon inducers including viral infections produce diminished responses upon re-exposure. It proved difficult to make interferon in sufficient quantities for trials due to the limitations of human tissue culture techniques. Recently, recombinant DNA technology has been employed as an alternative method of producing large quantities of interferon in prokaryotic (bacteria) cells. In this technique, genes for human interferon are inserted into E. coli genome. The interferon thus produced could be used to perform extensive clinical trials against a variety of human viral infections. The major drawbacks of interferon therapy are:
- Interferons will have to be obtained from human subjects if it is to be used in the treatment of human viral disease.
- In human subjects, its serum half-life is not more than 15 – 20 minutes. The brief survival of interferon in the body may be attributed to its rapid distribution and excretion from the body.
- Numerous clinical trials of interferon utility suggested that it is not as useful in the therapy of viral infections as initially suspected.
(7) Miscellaneous Agents
(i) Ureas and thioureas: Members of this class exhibit good antiviral activity but they could not be evaluated further because of their immunosuppresive side effects.
(ii) Guanidines and biguanidines: Guanidine hydrochloride shows selective inhibition of enteroviruses and more specifically polio viruses. But probably due to its rapid urinary excretion, the drug failed to achieve clinical status. The mode of action involves the selective inhibition of viral RNA polymerases. An extensive investigation in this class is still going on.
(iii) Heterocyclic dyes: The members of this series having antiviral potential includes proflavin and neutral red. The herpetic lesions are painted with 0.1% solution of these dyes and then the painted area is exposed to 16 – 30 W fluorescent light for 15 minutes. This results in the breakdown of viral DNA and the death of the virus. The painted area may be re-exposed to the light at the interval of 6 – 8 hours.
(iv) Gamma globulin (Immuno globulin): It is the antibody-rich fraction of the plasma which contains a variety of antiviral antibodies and is thought to act by preventing the penetration of the virus into the host’s cell. It is orally ineffective. After a single injection, it offers protection against viral infection for about 2 – 3 weeks.
These immunoglobulins are derived from β lymphocytes and plasma cells in very low amounts. Usually, only one class of immunoglobulins is initially synthesized by a given immunocyte and a clone of immunocytes synthesizes a specific type of immuno globulins.
Details about the fate of immunoglobulins are not available. The liver serves as a major site where they are degraded. Rarely immunoglobulines eliminated through the urine.
(v) Antibiotics: Several antibiotics of diversified structures have demonstrated antiviral activity to less or more extent. Extensive studies in this regard are yet to be made in this field. Examples of antibiotics possessing significant antiviral activity include rifamycin, bleomycin, gliotoxins, and clindamycin A. However, the doses for the antiviral activity of these antibiotics are usually higher than the dose needed for antibacterial activity. Due to this reason, the use of these drugs as antiviral agents is always accompanied by a high risk of adverse effects.
The antiviral activity of these antibiotics may be due to their ability to inhibit:
- assembling of particles into a mature virion, and
- viral envelope formation.
Various polymerase enzymes involved in either RNA and DNA synthesis in viruses are the principal target sites of action of these antibiotics.
(vi) Idoxuridine: During the next decade, in 1959 Idoxuridine, another antiviral agent was discovered, by Prusoff and actively used against the vaccine herpes virus. Idoxuridine (Herpid, Kerecid) competes with thymidine during the synthesis of DNA. It interferes with the multiplication of the herpes simplex virus which needs DNA, but, of course, it also interferes with mammalian cells, especially bone marrow.
In the form of a solution in dimethyl sulphoxide, idoxuridine penetrates the skin and can be effective by topical application in cutaneous herpes zoster.
(vii) Other investigational antiviral agents: These include phosphonoformic acid, bromovinyldeoxyuridine, 2-(3, 4-dichloro phenoxy)-5-nitrobenzenitrile and 4, 6- dichloroflavan. The former two agents are found to be effective against herpes virus infections while the latter two are effective against infections caused by enteroviruses or rhinoviruses.
Make sure you also check our other amazing Article on : What are Anti-Leprotic Agents?